Inventum.AI —
AI solutions for Drug Discovery
Design molecules in just 4 clicks and generate structures in 72 hours instead of weeks.
in vitro validated
40%
cost reduction at the early stages
by 5 times
acceleration of R&D processes
25%
increase in prediction accuracy
AI-driven Drug Discovery
with Experimental Validation
Our platform leverages cutting-edge AI to accelerate molecular design, covering ligand generation, binding site identification, molecular docking, scaffold growth, and interaction analysis. Backed by in vitro validation, Inventum.AI ensures real-world applicability and reliability, delivering precise affinity and ADMET predictions — all within an intuitive, user-friendly interface.
“Generating Targeted Molecular Structures Tailored
to Medicinal Chemists' Needs”
— this is our mission. How do we make it a reality?
Derived from Natural Interactions in Existing Protein-Ligand Complexes
Our methodology leverages nature-inspired, empirically-derived data, employing high-resolution crystallographic structures to train our Generative AI. This approach faithfully reproduces molecular recognition principles as observed in natural systems, thus reducing dependency on artificial parameters and enhancing the prediction of biologically relevant binding interactions.
Read the article
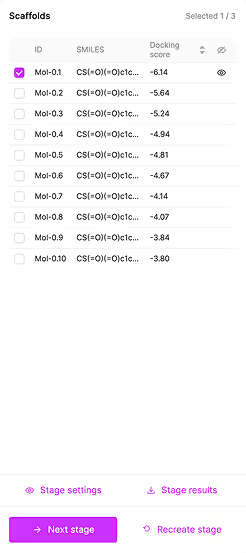
Step-by-Step Generation Process
- Positioning the selected starting fragment.
- Generating the scaffold.
- Generating the periphery.
With the ability to select chemotypes of interest at each stage. It allow us to direct the progress of generation towards the most interesting chemotypes.
The Best Search of Binding Sites
Our AI-based binding site search model, SiteRadar, has demonstrated superior performance over existing solutions in terms of binding site prediction accuracy.
Read the article
Novelty
Our models meticulously construct molecular structures atom by atom, enabling access to a diverse chemical space.
High Specificity
Our methodology employs optimized atom types within macro-molecular environments, fostering the generation of structures that exhibit strong complementarity with the ligand-protein binding site.
Drug-likeness
We use proprietary criteria to evaluate the drug-likeness of our generated structures, ensuring compliance with up-to-date medicinal chemistry standards.
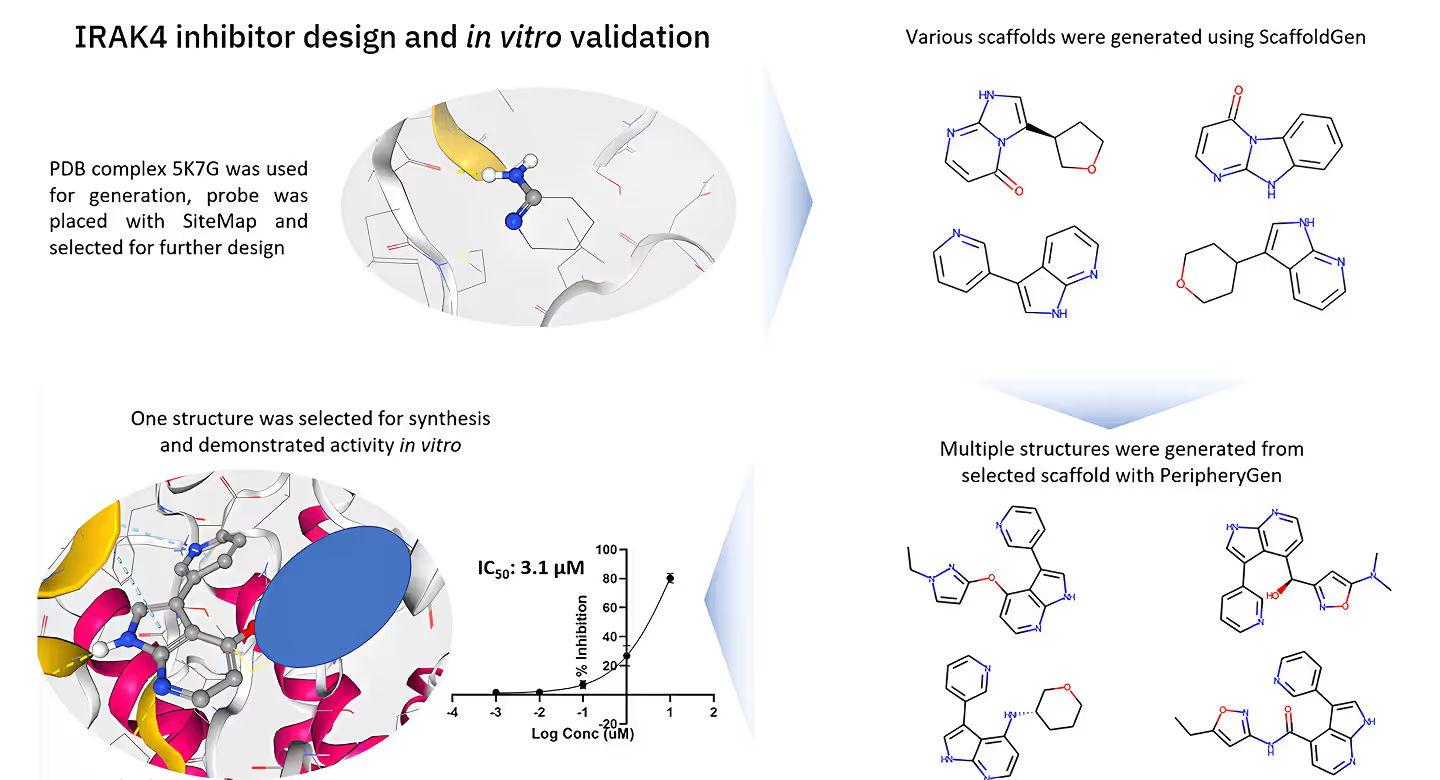
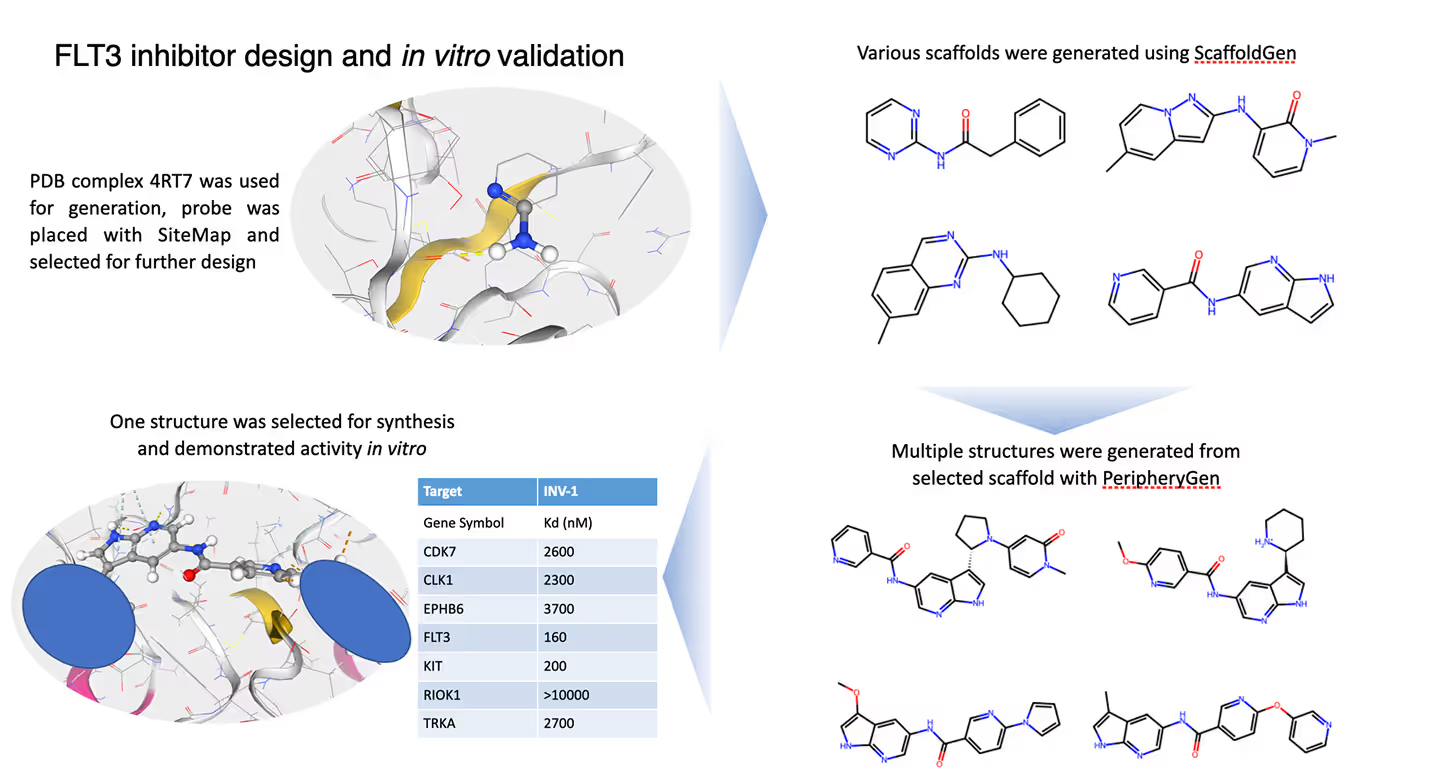
Experimentally Validated
Our platform is supported by robust experimental validation. All molecules are rigorously tested in vitro to confirm their efficacy and safety.
NBG Algorithm
Ligand structures are generated atom by atom to complement the macromolecular environment. Optimal atom types are selected by analyzing the macromolecular context using deep learning models trained on crystallographic data.
In silico Validation
NBG successfully reproduced the crystal structure of the CDK2 inhibitor, demonstrating strong correlation with the established Glide (Schrödinger) scoring function.
We are Transforming the Future of Drug Discovery
